How to Report Your Electroconvulsive Therapy (ECT) Injury to the FDA

This post explains what adverse medical event reporting is, why it matters, what keeps ECT -harmed patients from reporting injuries to the FDA, and how to submit yours using MedWatch.
This post contains a lot of useful information, organized into sections with links and a table of contents for easy navigation.
You can read the whole article or click on one of the headings below to skip ahead to a section that interests you.
Quick Start Links
- Key Takeaways
- Written Instructions
- Video Tutorial
- Slideshow Tutorial
- Reporting guide download
- Find my report
All Links
Key Takeaways
- It’s important to report injuries from ECT because it helps regulators keep track of how often serious problems occur. It also helps them identify previously unknown risks.
- When the FDA receives enough reports, it will investigate and take action to protect patients from unknowingly risking injury.
- While the form looks overwhelming, only four questions need to be answered, and can be done without your medical file.
- It’s better to report what information you have than none at all.
- Reporting adverse medical events is voluntary for patients. Many ECT patients and their families are unaware of how to report ECT injuries. Some patients are too injured to report injuries without help from family, caregivers, mental health professionals, or patient safety advocates.
- Doctors and nurses treating patients with a medical device are obligated by their code of ethics to report serious adverse effects caused by a medical device. Medical professionals treating patients with a history of ECT are also ethically responsible for reporting suspected ECT injury.
- Device manufacturers are legally required to follow up on MAUDE reports to better understand what happened so that risk of future injury can be reduced or eliminated.
- During court depositions taken for the Riera v Somatics, LLC case, The CEO of the Thymatron medical device testified that Somatics in more than 50 years of making ECT devices, the company had never followed up on any adverse event reports filed with the FDA, willfully negligent of their responsibility to reduce risks and ensure patient safety.
- The successful settlement of Riera v Somatics, LLC proves how not following up on ECT patient reports can be used to demonstrate willful negligence.
- Doctors, ECT providers, mental health counselors, and social workers are bound by their code of ethics to report the suspected harm of individuals with disabilities—even if it was accidental. Failure to report the injury of an individual from a vulnerable population (living with disability) is professional negligence.
What is Adverse Medical Event Reporting
Adverse medical event reporting is a way to monitor how people react to new medications and medical devices. This helps researchers identify any previously unknown negative reactions or outcomes.
What is MedWatch
“MedWatch, the FDA’s medical product safety reporting program for health professionals, patients and consumers.”
“Health professionals, consumers and patients can voluntarily report observed or suspected adverse events for human medical products to FDA. Voluntary reporting can help FDA identify unknown risk for approved medical products.”
“MedWatch receives reports from the public and when appropriate, publishes safety alerts for FDA-regulated products such as:
–Prescription and over-the-counter medicines
–Biologics such as blood components, blood/plasma derivatives and gene therapies.
–Medical devices such as hearing aids breast pumps, and pacemakers.
–Combination products such as pre-filled drug syringe, metered-dose inhalers and nasal spray.
–Special nutritional products such as dietary supplements, medical foods and infant formulas.
–Cosmetics such as moisturizers, makeup, shampoos, hair dyes and tattoos.
–Food such as beverages and ingredients added to foods.”
MedWatch: The FDA Safety Information and Adverse Event Reporting Program | FDA
Who Can Report to MedWatch
People from all countries can submit adverse medical reports to MedWatch.
Is MedWatch Confidential
“Reporting to MedWatch is easy, confidential, and secure.”
FDA 101: How to Use the Consumer Complaint System and MedWatch | FDA
“The FDA recognizes that the confidentiality of the identities of both providers who report adverse events and patients is an important concern of health professionals. To encourage reporting, the FDA carefully protects the identities of providers who report and patients contained in FDA records and will not release such information to the public.”
Introducing MedWatch (fda.gov)
What is the MAUDE database
The Manufacturer and User Facility Device Experience (MAUDE) contains medical device adverse reports. This is where your anonymous adverse report will be stored.
“MAUDE data represents reports of adverse events involving medical devices. The download data files consist of voluntary reports since June 1993, user facility reports since 1991, distributor reports since 1993, and manufacturer reports since August 1996. The searchable database data contains the last 10 year’s data.”
Manufacturer and User Facility Device Experience Database – (MAUDE) | FDA
Device Manufactures, Importers, and Device User facilities Must Report Adverse Events
“Manufacturers and importers must submit reports when they become aware of information that reasonably suggests that one of their marketed devices may have caused or contributed to a death or serious injury or has malfunctioned and the malfunction of the device or a similar device that they market would be likely to cause or contribute to a death or serious injury if the malfunction were to recur.
Manufacturers must send reports of such deaths, serious injuries and malfunctions to the FDA.
Importers must send reports of deaths and serious injuries to the FDA and the manufacturer, and reports of malfunctions to the manufacturer.”
“Device user facilities must submit reports when they become aware of information that reasonably suggests that a device may have caused or contributed to a death or serious injury of a patient in their facility.
Death reports must be sent to the FDA and the manufacturer, if known. Serious injury reports must be sent to the manufacturer or to the FDA, if the manufacturer is not known.”
MAUDE – Manufacturer and User Facility Device Experience (fda.gov)
Medical Providers Have an Ethical Obligation to Report Adverse Events
“A physician who suspects that an adverse reaction to a drug or medical device has occurred has an ethical responsibility to:
1. Communicate that information to the professional community through established reporting mechanisms.
2. Promptly report serious adverse events requiring hospitalization, death, or medical or surgical intervention to the appropriate regulatory agency.“
Required Reporting of Adverse Events | American Medical Association (ama-assn.org)
The MAUDE Database’s Role In Litigation
The MAUDE Database has proven itself useful in recent ECT litigation; device manufacturers ignored their legal duty to file adverse medical reports:
“A reasonable jury could find that the ECT device manufacturer caused Plaintiffs’ brain damage through failure to warn their treating physicians of brain injury, or alternatively by failing to investigate and report allegations of brain damage and permanent memory loss to the FDA, so that information would be available to the public.
The duty to “investigate and report” allegations of “serious injury or death” by the manufacturer has resulted in ZERO reported adverse events for over four decades, demonstrating a conscious disregard to comply with reporting obligations.”
ECT Lawsuit Victory – Riera v. Somatics, LLC, 2018
A Successful FDA Reporting Campaign
The nonprofit, Benzo Coalition, was successful in getting a black box warning put on benzodiazepine leaflets by encouraging those harmed to report their injuries to the FDA.
- FDA 2020 Benzo Boxed Warning Freedom of Information Act – Benzodiazepine Information Coalition (benzoinfo.com)
- FDA requiring Boxed Warning updated to improve safe use of benzodiazepine drug class | FDA
Imagine what we can do with ECT!
What keeps Injured ECT Patients From Reporting
Common reasons ECT patients don’t report are privacy concerns, cognitive disability, missing or forgotten information, and trauma.
Let’s break down these barriers:
Fear of retaliation from the device manufacturer or doctors.
MedWatch is anonymous. You can choose to keep your identity private from the device manufacturer (form section: reporter> name & privacy).
The form is confusing!
Yes, the form can be especially confusing for former ECT patients.
Our guide will walk you through the entire reporting process.
I don’t have all the information they ask for.
The good news is only four questions need to be answered and the information doesn’t have to be super specific. You don’t have to prove your injuries– only report them.
Remember, it’s better to report a small amount of information rather than nothing.
What’s the point? The FDA doesn’t care about us.
There’s no doubt the FDA has betrayed the public with how its managed the ECT device:
- The FDA’s Regulation of ECT (Shock Treatment): A Beginner (or Refresher) Course
- Comments by Shock Survivors and Their Loved Ones – Mad In America
- Open Letter of Appeal to FDA about Electroconvulsive Therapy (ECT)
- Open Letter to the FDA’s Denial of ECT’s Harms – 2021
- Appealing the FDA’s Denial of ECT’s Harms
That said, the agency’s behavior shouldn’t be a reason to give up your right to be heard and participate in the regulatory process.
I’ve tried reporting my injuries before. I’m sick of no one listening.
I understand how traumatic trying to get help after ECT is. Knowing the limits of reporting beforehand can make this easier.
It won’t get me the help I need now.
It’s true. Submitting a report doesn’t guarantee immediate or future help for us, but its a crucial part of the process:
- It serves as a stepping stone to investigations and safety actions.
- It’s a way to be seen and heard in a formally recognized way.
- It can lead to beneficial changes for past, present, and future patients.
- Even if the FDA doesn’t follow through, reports stored in the MAUDE database can be used in court as evidence and in research.
Speaking truth to power:
As we have seen, ECT device manufacturers have neglected their legal and ethical duties to report adverse events to the FDA.
This negligence has contributed to scores of ruined lives.
Reporting is just one way to make our injuries known and protect future patients.
Even a little effort can go a long way.
Hope From the Past; Ten ECT Patients Change The World
In 1982, the electroshock device almost got a free pass to class II risk classification.
An ECT survivor named Marilyn Rice rallied ten peers to speak at the FDA reclassification hearings. By speaking out this small group protected countless patients by ensuring the ECT device stayed in the highest risk category for decades.
“It has been said that it only takes a few people to change the world. Rice had understood this from the beginning. She had worked hard to get survivors to testify at the FDA because she knew that if they did not, the FDA would grant the APAs petition. She was right. After the public hearing ended, when the panel met for closed discussion and to vote, it was clear that some of the panel members were deeply concerned about the testimony from former patients, and could not scientifically or in good conscience reconcile what they had heard with a vote for Class II.”
Doctors of Deception: What They Don’t Want You to Know about Shock Treatment by Linda Andre
This victory shows us what a handful of ECT survivors can pull off when they work through the system.
It’s our turn to stand together and be heard.
What Reporting is Like
I’ve known about the MAUDE database since 2012 but had several hang-ups that kept me from reporting.
The form was confusing.
Years of being denied help for my brain damage broke me emotionally.
I was done trying to get help for my injuries.
Not that it mattered– I couldn’t get my medical records so I figured I didn’t have enough information to file a report, anyways.
I also worried about my privacy and possible retaliation from the device manufacturer, doctors, and hospitals.
I doubted my report would be taken seriously after learning how the FDA has failed to protect the public in regard to ECT for as long as its overseen the device.
Then a few years ago a friend told me how adverse reporting worked. They showed me it was easier than it looked, and while it might not get me what I need right now it was still worth reporting my injuries to the FDA.
Knowing what I could expect and that my submission was confidential alleviated my anxieties.
After that, the hardest part was remembering details and writing out my injuries within the word count.
Using the writing strategies included below helped simplify and speed up the process.
What Should You Report to MedWatch
“What information do I need to complete a report?”
“To submit a report, the reporter should have following information at minimum:
–Description of the adverse event or problem that occurred
–Name of the suspect medical product if reporting about a prescription or over-the-counter medicines*, biologics, special nutritional products, cosmetics, food etc.
–Name of the suspect device if reporting about a medical device (including diabetes glucose-test kit, hearing aids, breast pumps, and many more products)
–Reporter’s name (FDA will not disclose your identity to the manufacturer if requested in the form.)
*Other details such as National Drug Code or NDC number, lot number, and expiration date should be reported when available but are not required to submit a report.”
MedWatch Online Voluntary Reporting Form (fda.gov)
Reporting Guide Download
Tips:
If you have trouble holding information in your mind (working memory) I recommend printing the guide so you can circle or highlight symptoms you might have before writing your experience.
It can also be used as a reference while you write and submit your report so you don’t have to switch back and forth between windows while you work.
SlideShow
How to Report Unexpected Problems After ECT to the FDAVideo
Note: these instructions are for filling out the form with the least amount of information possible.
If you do have more information beyond the required questions, feel free to answer as many questions as you feel comfortable with. Good luck!
Written Instructions
- Visit the MedWatch website MedWatch Online Voluntary Reporting Form (fda.gov) URL: https://www.accessdata.fda.gov/scripts/medwatch/index.cfm
- Select the red button that says “report a problem”
- Select Consumer/Patient (FDA Form 3500B) button
- PROBLEM PAGE Tell us what happened section: enter details describing what happened. See tips if unsure what to write.
- PROBLEM PAGE, CONTINUED select the box “for problems with a medical device”
- On DEVICE PAGE enter the name of the medical device: ECT machine. (You don’t need a specific name or model, just device type)
- On the REPORTER PAGE enter your first and last name
- Optional: check the box if you DON’T want the device manufacturer to contact you.
- Review your answers & click submit.
- You’re done!
- Please forward your confirmation email to contact[at]lifeafterECT[dot]com so we can track submission numbers.
What Happens Next
According to the FDA when you submit your report:
“FDA staff enter the report into a database so that it is available for review and comparison to other reports.
-An FDA safety evaluator, often a pharmacist, doctor, or nurse, reviews the report and examines the database for similar reports.
-The FDA monitors the data for trends and conducts an investigation, if appropriate.
-The FDA takes necessary action to protect public health.
FDA actions may include:
–issuing safety alerts with recommendations to monitor a product’s use, adjust the way it is used, or stop using it
–updating the product labeling to reflect new warnings
-inspecting the manufacturer
–requiring a product to have a Medication Guide—a consumer-friendly instruction sheet provided to patients each time they fill a prescription to help them use the drug safely
–requesting a change in the product’s design, manufacturing process, packaging, or distribution
–requesting a company to recall a product
–requiring a manufacturer to conduct further studies to demonstrate the product’s safety prior to allowing the product back on the market“
FDA 101: How to Use the Consumer Complaint System and MedWatch | FDA
After the Report Has Been Reviewed
I received this email thanking me for my submission:
Dear Reporter:
Thank you for submitting your report to MedWatch, the FDA Safety Information and Adverse Event Reporting Program.This acknowledgment confirms that your report was received.
Reports are added to a post marketing safety database with similar reports and reviewed by the FDA’s post marketing safety staff. Voluntary reports are essential for ensuring the continued safety of FDA-regulated products. One or two well documented case reports may provide an early signal of unexpected problems and lead to additional evaluation. This may result in FDA regulatory actions that improve the safety of the products used in patient care each day.
You will only be contacted for follow-up by FDA if additional information on this report is needed in our evaluation process.
Again, thank you for taking the time to submit your report. If you have any questions, please send them to: [email protected] .
Sincerely yours,
FDA MedWatch Team
Over a month later I received another email explaining what would happen to my submission and an access number I can use to view my anonymized report in the MAUDE database.

Dear Reporter:
Thank you for submitting your Medical Device Report (MDR) to the Food and Drug Administration’s (FDA) MedWatch program ( http://www.fda.gov/medwatch/ ) FDA’s Center for Devices and Radiological (CDRH) is responsible for reviewing MDRs.
Your report will be entered into CDRH’s Manufacturer and User Facility Device Event (MAUDE) database. CDRH’s Office of Regulatory Programs (ORP) monitors the MAUDE database to identify device problems and to determine trends. If necessary, you will be contacted to obtain further information.
ORP conducts educational outreach, publishes safety alerts and public advisories and recommends product recalls if necessary. ORP also uses MDR data to disseminate information to the health care community via medical/health publications.
MDR data is also used for presentations, field training, for educating manufacturers, hospitals and other health care facilities and for major medical professional and health meetings.
All MDR reports entered into the MAUDE database and edited to remove identifying information are on the Internet at http://www.fda.gov/cdrh/maude.html.
We do not provide individual feedback for reports received, however, please note our appreciation and be assured that our scientific staff will review the information submitted.
Please reference your access number below, if you would like to file a follow-up report, wish to correspond further or if you have any questions about your report.
Access Number: [REDACTED]
Thank you again for participating in the MedWatch program.
Sincerely,
Michelle Rios, MS
Assistant Director
Medical Device Reporting (MDR Team)
Division of Regulatory Program 3 | Office of Regulatory Programs
Office of Product Evaluation and Quality
Finding Your Report in The MAUDE Database
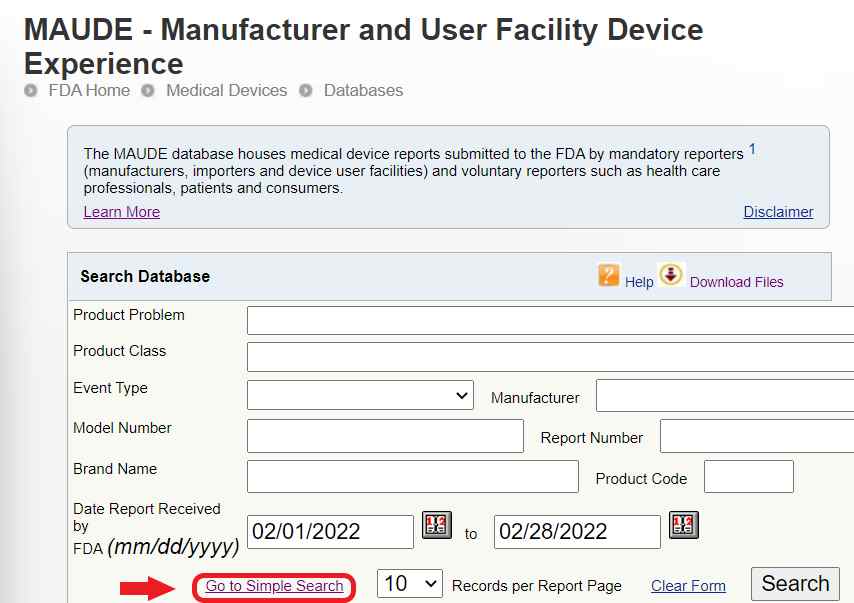

After submitting your report you should get a confirmation email thanking you for your submission.
Eventually, you’ll get a 2nd email with an access number to view your anonymized report in the MAUDE database.
- Visit MAUDE – Manufacturer and User Facility Device Experience (fda.gov) I recommend using the “simple search” setting (the above link is set to simple search)
- Copy/Paste your Access Number in the Search Database box.
Select the year you submitted your report or “ALL YEARS.” - Click “search”
- A link to your anonymized report should appear.



