Edit 1/7/23, error with date of in 2007 Maeve A. Mangaoang and Jim V. Lucey published Cognitive rehabilitation: assessment and treatment of persistent memory impairments following ECT
Glossary
- Adverse medical reporting is “the act of monitoring a medication or medical device after it has gone to market to identify and evaluate previously unknown adverse reactions or outcomes.” source.
- Food and Drug Administration (FDA) “The United States Food and Drug Administration is a federal agency of the Department of Health and Human Services.” source.
- The Manufacturer and User Facility Device Experience (MAUDE) database is where adverse medical reports submitted to the FDA are stored.
- “Manufacturer and User Facility Device Experience (MAUDE) database represents reports of adverse events involving medical devices. The searchable database data contains the last 10 years of medical device report (MDR) data.” source.
- Medical Device Amendments of 1976 (MDA) “Intended to provide reasonable assurance of the safety and effectiveness of medical devices” source.
- MedWatch The FDA program where adverse reports are submitted. “the FDA’s medical product safety reporting program for health professionals, patients, and consumers.” source.
- Safe Medical Devices Act of 1990 (SMDA) “Safe Medical Devices Act of 1990 – Amends the Federal Food, Drug, and Cosmetic Act (FDCA) to require medical device user facilities to report to the Secretary of Health and Human Services, the manufacturer, or both whenever they believe there is a probability that a medical device has caused or contributed to a death, illness, or injury.” source.
ECT ADVERSE REPORTING UPDATE
This spring, Life After ECT launched a comprehensive FDA reporting guide for injured electroconvulsive therapy (ECT) recipients. This post is a progress update and an overview of our future goals.
THANK YOU
First, thank you to everyone who has already submitted their report! We sincerely appreciate your efforts.
ECT ADVERSE REPORT STATISTICS
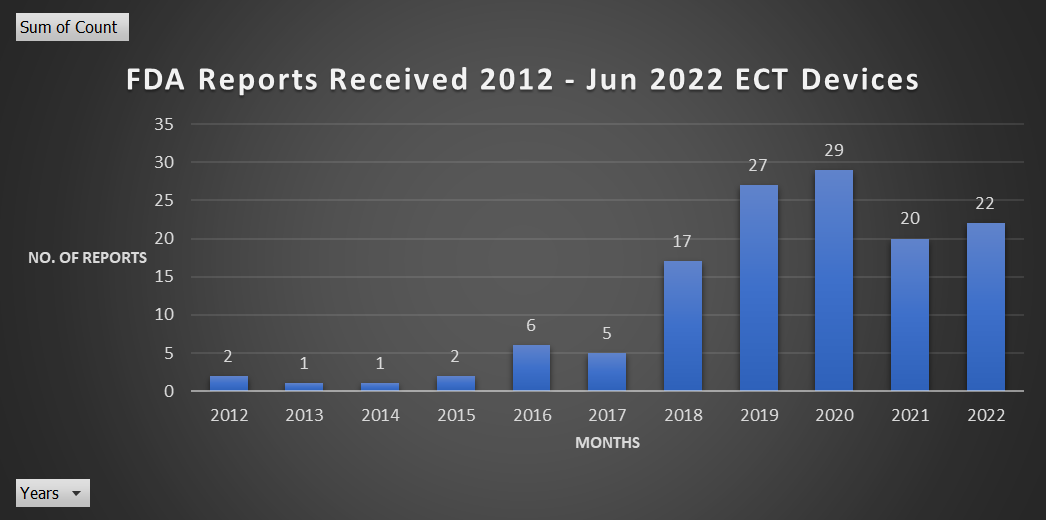
Available ECT adverse report data through the Manufacturer and User Facility Device Experience (Maude) database begins in 2012.
Below is a breakdown of adverse ECT reports to the FDA found in the MAUDE database:
- All-time reports – 132 reports since 2012.
- The highest number of reports in a year – 2020, with 29 reports.
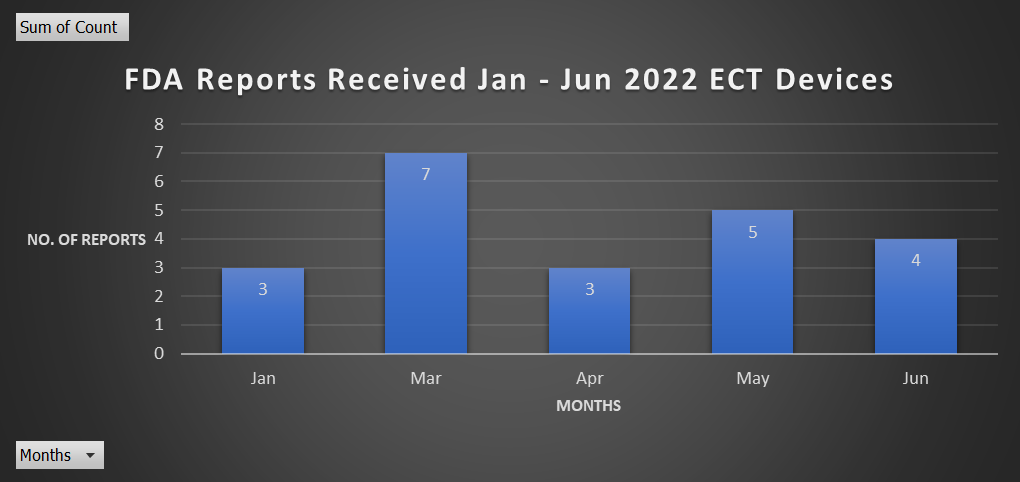
- This year, 2022, from January through August, there have been 22 reports.
All Time Reports, 2012 – 2022
Reports January – June 2022
HOW MANY RECEIVE ECT VS. ADVERSE REPORTS SUBMITTED
We don’t know how many people receive ECT because these statistics aren’t tracked.
What is known:
ECT proponent and researcher Harold Sackeim testified under oath in 2006 that 2 million people worldwide receive ECT:
Sackeim says 2 million a year have ECT – YouTube
There’s been an increase in US hospitals providing ECT.
From the Psychiatric Times article, ECT: Dangers on Either Side of the Pond:
“Given ECT’s national reimbursement practices, it is unsurprising that the Substance Abuse and Mental Health Services Administration’s National Directory of Mental Health Treatment Facilities ECT provider list jumped from 335 clinics in 2018 to 449 in 2020. The 34% increase in US hospitals providing ECT since device reclassification may reflect what happens when hospitals identify an unregulated income source.
Sarah Price Hancock, ECT: Dangers on Either Side of the Pond, 2021
Despite Sackiem’s sizable estimate of global annual ECT recipients and a known increase in the US, few doctors or recipients report adverse events to the FDA.
Some might argue bad outcomes have been reduced due to refinements in ECT devices and treatment methods; however, medical research and other injury reports tell a different story.
ABSENT DOCTOR REPORTS
Medical device-related adverse events are commonly underreported across specialties:
“It is difficult to accurately determine the rate of under-reporting of medical device adverse events because the actual number of incidents is unknown. However, the case studies in this review are in agreement with the FDA estimate of a reporting rate of 0.5%.”
Adverse reports from ECT providers are astonishingly low.
ECT is no exception; these sparse numbers are unusual considering the commonality of serious adverse events in ECT, many of which are documented for research & educational purposes.
Peruse any online medical journal, and you’ll find dozens of case reports of unusual or deadly ECT incidents. Many American electroshock providers are publishing these adverse events in journals but not relaying them to the FDA, even though it’s mandatory.
According to the MAUDE Database website:
“Device user facilities include hospitals, outpatient diagnostic or treatment facilities, nursing homes and ambulatory surgical facilities. Device user facilities must submit reports when they become aware of information that reasonably suggests that a device may have caused or contributed to a death or serious injury of a patient in their facility. Death reports must be sent to the FDA and the manufacturer, if known. Serious injury reports must be sent to the manufacturer or to the FDA if the manufacturer is not known.”
MAUDE – Manufacturer and User Facility Device Experience (fda.gov)
Common cardiac risks should increase adverse reports.
Serious adverse events like bradycardia (slowed heart rate) and asystole (heart attack) are common in ECT according to a letter to the editor in The Journal of ECT titled The Trigeminocardiac Reflex in Electroconvulsive Therapy:

“Acute poststimulus bradycardia and asystole during electroconvulsive therapy during ECT.”
The Trigeminocardiac Reflex in Electroconvulsive Therapy : The Journal of ECT
“Cardiovascular disease was the most common medical cause of death (n = 49, 40 %). An older age, a Charlson Comorbidity Index of 1 or more, atrial fibrillation, kidney disease, reflux disease, dementia, and cancer were associated with increased risk of death by medical causes.”
One would assume if such serious events happen frequently, there would be more adverse reports from user facilities. However, that’s not the case.
MAN DIES FROM SUDDEN CARDIAC DEATH AFTER ECT
The Journal of Psychiatry and Psychiatric Disorders published a case report in 2017 of an ECT-related death. A man with significant cardiac risks was referred for ECT. He would die from asystole following an electroconvulsive therapy session:
“On the day of the 14th treatment, the patient was prepared the same way as on all previous occasions. His EKG, vital signs, and serum electrolytes were all within normal limits. After administration of the bitemporal stimulus, the patient had a 15-second clinically visible response. After the treatment, he appeared to stabilize but then developed progressive bradycardia and, despite repeated doses of atropine and epinephrine the bradycardia ended up in asystole. External resuscitation was attempted, but after 45 minutes the patient was pronounced dead.”
Sudden Cardiac Death After ECT, Journal of Psychiatry and Psychiatric Disorders, 2017
This man’s death was never reported to the FDA.
I searched 2017 entries in the MAUDE database. Not a single report resembles this man’s death. To be thorough, I also checked 2016 submissions in case his death occurred a year before the case report was published. No asystole-related fatalities were reported.
Next, I checked the “death” section on the ECT device’s Total Product Life Cycle Report, a database that “integrates premarket and postmarket data about medical devices”. This search method yielded two death reports after 2016; The first was submitted by the ECT device maker, Mecta, in 2018 for a death that occurred in 2008.
The second, also from Mecta, relayed details of a report they received from an ECT provider:
“On (b)(6) 2016 email from distributor notifying us of death by myocardial infarction two hours after ect treatment.”
MAUDE Adverse Event Report: MECTA CORPORATION MECTA SPECTRUM 5000Q ECTDEVICE (fda.gov)
While details fit the case report, Mecta’s submission said the recipient died 2 hours post-ECT. Our case had complications close to the time of seizure induction, and he was dead 45 minutes after. These differences, coupled with the commonality of ECT-related cardiac complications, make it unlikely to be the same case.
Another unusual aspect of this: the death Mecta submitted wasn’t directly reported to the FDA by the user facility that treated the decedent. Instead, they contacted the manufacturer, Mecta, who later filed the report with the FDA.
Judging by the staggering absence of doctor reports, it’s more than most seem willing to do, but why not also relay this tragic outcome through “established reporting mechanisms,” as required?
MEDICAL PROVIDERS HAVE AN ETHICAL OBLIGATION TO REPORT ADVERSE EVENTS
“A physician who suspects that an adverse reaction to a drug or medical device has occurred has an ethical responsibility to:
1. Communicate that information to the professional community through established reporting mechanisms.
2. Promptly report serious adverse events requiring hospitalization, death, or medical or surgical intervention to the appropriate regulatory agency.“
Required Reporting of Adverse Events | American Medical Association (ama-assn.org)
Few ECT providers report adverse events.
I was disturbed to find so few ECT provider reports in the database; what entries I saw seemed primarily concerned with the ECT equipment:
“Pt here for electro-convulsive therapy treatment. When electroshock given, a spark and smoke arose from electropad on r temple of pt. Electro pad actually popped off on skin leaving a small black mark. Black mark wiped off when skin cleansed for examination and no obvious burn mark was noted. Electro-convulsive therapy device removed from service pending biomed review.”
MAUDE Adverse Event Report: SOMATICS, LLC THYMATRON IV DEVICE, ELECTROCONVULSIVE THERAPY (fda.gov)
Functioning equipment is vital for patient safety, so it’s great some issues get reported. However, ECT recipients’ submissions show us doctors are disregarding patient adverse reports caused by properly functioning shock machines.
WE KNOW UNEXPECTED SIDE EFFECTS ARE COMMON
Unexpected adverse reports regularly appear in news articles, blog posts, books, patient experience research, and mental health forums.
Examples:
Research
- A review of consumers’ perspectives on electro-convulsive therapy
- Recipients’ experience with information provision for electroconvulsive therapy (ECT)
- Women’s Experiences of Electro-convulsive therapy: A Qualitative Study
- Voices From Within: A Study of ECT and Patient Perceptions
- Voices of people who have received ECT (India)
Forums
- ECT in SANE forums
- ECT (Electroconvulsive therapy) | Mental Health Forum
- Has anyone used ECT Therapy for depression? | Mayo Clinic Connect
- The Depression Forums
Other media
- Comments by Shock Survivors and Their Loved Ones
- Electroconvulsive therapy malpractice cases
- My Experience With Electroconvulsive Therapy (ECT)
- Podcast interview Dr Susan Cunliffe: Escaping Psychiatry and ECT – A Physician’s Experience – Medical Error Interviews
- Psychologist’s memoir is a tale of misdiagnosis, mistreatment, and healing
- The Electroshock Quotationary
- What’s life like after Shock Treatment (ECT)?
Support groups:
As a support group admin with +500 members, I reviewed group applications and moderated frequent posts describing serious ECT side effects my peers weren’t warned of.
- See Electroconvulsive Therapy Side Effects, a list made in part from an informal ECT recipient survey.
ECT RECIPIENTS REPORT INJURIES
Those brave enough to seek help from the doctor who harmed them are told often their experience isn’t real, their mental illness is to blame, or ignores them.
These psychiatrists rarely consider these reports valid or their duty to relay these outcomes to the FDA.
MAUDE reports from ECT recipients:
“…i had 9 treatments done after being evaluated and educated about the process and was told that i would only have mild temporary memory loss. Which couldn’t be further from the truth, in (b)(6) 2016 immediately after my last treatment i started telling my provider about the severe headaches and vision loss but he ignored my issue. The issues continued to come and get worse and worse. I have a headache 24/7, vision issues, nausea/vomiting, severely painful muscles and joints, lost my memory that still has not return, completely forgot how to do my job, anger, agitation, increased suicidal thoughts, my depression has never improved whatsoever, dizziness, confusion, not able to focus or remember what i’m supposed to do day to day… My psychiatrist continues to deny that my treatments had anything to do with any of these other issues. I’ve also found out information about ect’s that was never told to me.”
“I endured 9 rounds of electroconvulsive therapy from 2010 to 2011. I had over a year of intense recovery and still have difficulty. I reported problems to my dr many times, no one believed that ect could have caused it.”
“Ect’s caused perm. Seizures, bi frontal brain atrophy. Shrinking of grey and white matter in brain, perm. Cognitive physical behavioral issues. Was (b)(6) and now can’t remember how to spell. The doctors kept telling my parents my memory and seizures and talk incomplete sentences would all be fine a few months after we stopped ects, but they kept doing them even after i have positive signs of brain damage shown in two eeg’s and frontal atrophy in frontal lobes.“
“I am a shell of who i was before. All of my cognitive / executive functioning has been negatively effected. I cannot control my emotion. I get flustered easily and it is humiliating. My memory is all messed up. Events as much as 2 to 3 years before are gone. I can still remember minute details of insignificant things but may forget a conversation i just had recently with someone. I get confused, always forget where i put things, cannot get things organized and live with piles of “stuff” all over my house…no drs believe me that ect caused this, so none have done any tests.“
ECT’s harms go undocumented in other ways.
Though not directly related to FDA adverse reports, these excerpts show how abuses, disabling injury, and death go unreported and undressed in other ways.
From the president of The Committee for Truth is Psychiatry, Linda Andre, to the FDA, 2010:
[emails from injured ECT recipients] were forwarded to me after being sent to Dr. Harold Sackeim of New York State Psychiatric Institute, the nation’s most prominent ECT researcher and, for the last 25 years, the principal investigator of an ongoing NIMH grant to study ECT’s cognitive effects. Dr. Sackeim had claimed that he had never seen a case of permanent anterograde amnesia and publicly offered to evaluate former patients who had experienced it…at least 195 such patients contacted Dr. Sackeim, consistently reporting the same serious adverse effects. Dr. Sackeim later testified that he had thrown away all communications from these patients and did not conduct any evaluations.”
Linda Andre, Committee for Truth in Psychiatry – Supplement Comment re FDA-2009-N-0392-1243
ECT proponent Max Fink stated in a 2012 interview that he’s observed abuses as a journal editor:
“I edited the Journal of ECT for the first 10 years. I read many submissions. And when you read them, you realize that people have . . . mistreated their patients. They don’t know it, but I know it from the way they write.”
He attributes this to a lack of standards, enforcement, and proper training:
Fink says delivery protocol standards need to be developed where they don’t exist and enforced where they do. “Very few physicians are trained properly (in ECT),” he says, adding that some institutions offer one-day training programs. He calls these programs “spurious” and “unethical.”
Max Fink, Shock, an investigation into the startling comeback of electroconvulsive therapy, page 27
From UK psychologist and professor John Read:
“At a staff meeting in my very first job as a clinical psychologist in the UK, I raised the issue of a man who had died on the ECT table the day before. I still recall the exact response of the psychiatrist: ‘That is none of your business and I am personally insulted by your insinuation that we killed him.’ When I pointed out that the man’s notes included ‘ECT contraindicated – serious heart condition’, I was evicted from the meeting – physically. A colleague and I had copied that page of the notes, accurately predicting that it would quickly be removed from the file. I tried for two years to get the hospital, professional and governmental authorities to investigate. I failed.”
Why is electroshock therapy still a mainstay of psychiatry? | Aeon Essays
From Dennis Cauchon, USA TODAY, 1995:
“Doctors rarely report shock treatment on death certificates, even when the connection seems apparent and death certificate instructions clearly indicate it should be listed.”
Patients Often Aren’t Informed of Danger of ECT | HealthyPlace
A recent example of a death not attributed to ECT; the coroner’s report said the person having post-ECT seizures died of a “heart attack in schizophrenia.”
We know doctors rarely refer ECT recipients for cognitive testing.
Lack of referrals seems due in part to ignorance and fear of liability, according to Robertson & Pryor in Advances in Psychiatric Treatment (2006):
“When individuals who have had ECT report ongoing memory disability, it is necessary for a clinician trained in neuropsychological evaluation to tease out the roles played by attention, concentration, overall slowed mental processing and deficits of executive function such as inability to shift mental set. The ECT psychiatrist and treatment team may not be trained in neuropsychological evaluation, since outside of research settings it is not routinely performed on people who have had ECT. When it is, it is usually initiated by the patient, not the doctor. Because of this, the treating psychiatrist may fear personal liability and thus be unwilling to attribute deficits to ECT.”
Many psychiatrists fought to keep neuropsychological testing from becoming a standard for ECT recipients.
In another corner of the FDA, you can see a concerted effort among individual psychiatrists and psychiatric organizations to weaken or eliminate risk information, safety measures, and testing provisions in the 2016 ECT device reclassification draft guidance docket.
Examples:
- the American Academy of Child Adolescent Psychiatry docket submission
- Comment from Max Fink FDA-2014-D-1318-0081_attachment_1.pdf
- Comments from Richard Abrams M.D. contraindations FDA-2014-D-1318-0159 , Software Life Cycle and Risk Management, intended use FDA-2014-D-1318-0250
- Comment FDA-2014-D-1318-0131
- comment FDA-2014-D-1318-0043
The APA fought against neuropsychological testing.
The American Psychiatric Association (APA), an organization representing 36,000+ psychiatrists gave similar feedback to the FDA on draft guidance for the proposed device reclassification.
- See Electroconvulsive Therapy (ECT) Devices for Class II Intended Uses: Draft Guidance for Industry, Clinicians and FDA Staff; Availability, FDA-2014-D-1318-0001
Their submissions requested the inclusion of more groups in the class II reclassification, like children and people with schizophrenia. They also requested various labeling be modified or deleted from the draft guidance.
From the APA:
“The APA does suggest several modifications to the wording in the proposed labelling. Detailed formal neuropsychological assessment [49] is not necessary in all patients treated with ECT and will not be possible in many treatment centers, resulting in a barrier to ECT treatment if this is required.”
“Thus, the APA suggests that the phrase “formal neuropsychological assessment” be deleted from the labelling. Instead, we recommend a staged approach to cognitive testing similar to what is used in other disorders in health care.”
“Screening assessments covering orientation, attention, and memory in all patients prior to and during ECT would permit identification of those individuals who may require more detailed testing. Asking patients and family members about subjective concerns about cognition during the ECT course can also identify individuals who may require more detailed testing or adjustments in ECT treatment parameters.”
https://www.regulations.gov/comment/FDA-2014-D-1318-0216
Laura Fochtmann, Professor of Psychiatry at Stony Brook University School of Medicine made similar arguments:
“Delete the recommendation for “formal neuropsychological assessment” from the labeling. A staged approach to testing is similar to the approach used in other aspects of health care in which screening for abnormalities in orientation, attention, and memory can be followed up with more detailed assessments as necessary.”
“Other medical treatments for serious illness are known to produce cognitive effects (e.g., cancer chemotherapies); yet formal neuropsychological assessment is not the norm before initiating these treatments. Furthermore, many patients would be unable to complete a formal battery of neuropsychological tests because of the severity of their psychiatric symptoms.”
“Requiring formal neuropsychological assessment would also create barriers to the availability and timeliness of care in that such assessments are costly and difficult to access.”
https://www.regulations.gov/comment/FDA-2014-D-1318-0216
Trust us. We’re doctors.
Rather than seek ways to meet this new standard that would ensure injured recipients access to rehabilitation, social services, and disability pensions if they become unemployable, the APA advocated against it. They didn’t list alternative tests in their letter to the FDA. They could be referring to the Mini-Mental State Exam (MMSE), a test commonly given to ECT recipients which interestingly, can be passed by lobotomized patients.
Not that it matters; without concrete standards and regulations, the APA can provide no assurances that its members will now believe harmed ECT patients and provide appropriate testing, rehabilitation, and treatment modifications when the need arises.
Eighty+ years in use, and it’s business as usual.
Nothing has changed for those harmed by ECT. Scant research on post-ECT rehabilitation exists. You’re guaranteed to find more studies on ECT given in high-risk groups to prove it’s safe for that population than you will on successful post-electroshock rehabilitation research.
Rehabilitation isn’t a priority.
In 2007 Maeve A. Mangaoang and Jim V. Lucey published Cognitive rehabilitation: assessment and treatment of persistent memory impairments following ECT:
“At the time of writing, no attempts have been made to rehabilitate patients who experience persistent adverse cognitive effects, but clinicians should be aware of the potential beneficial role of cognitive rehabilitation in the treatment and management of these effects.”
It’s 2022, and I still see frequent posts from injured recipients struggling to obtain testing that leads to rehabilitation.
Some, like me, were only able to get tested for unrelated reasons. In my case, I spent five years trying to convince my doctor that ECT damaged my brain. He refused to consider it. I was only able to get an evaluation when I went back to school to earn my GED; Brain damage was less compelling to my doctor than educational accommodations, it seems.
Formal assessments aren’t believed.
The lucky few who are evaluated often have our results interpreted by specialists who often refuse to attribute our deficits to ECT. Instead, they often suggest our mental illness is the cause of our cognitive impairments. This creates more hurdles to accessing rehab. Another example before I could get a referral to a dementia specialist I had to see a counselor to help me manage my “anxiety” the testing psychologist said was causing my working memory deficit.
My neuropsychological evaluation showed I have chronic encephalopathy and other major deficits common with head traumas. When I finally met with the dementia specialist, I was told, “ECT doesn’t cause the problems I was experiencing” and “there’s no need for more testing or treatment.” In a subsequent follow-up with my neurologist’s P.A., she said, “Well, we don’t know why you’re this way, but we know it’s not the ECT!”
DEVICE MAKER’S THOUGHTS ON THEIR RESPONSIBILITY TO HARMED ECT RECIPIENTS
On page 18 of a complaint filed with the Central District of California on September 11, 2017, Jose Riera; vs. Mecta Corporate; Somatics LLC, gives us insights into the company’s thoughts on those who allege brain damage from their product:
The CEO of MECTA, Ms. Robin Nicol, admits that these lawsuits alleged that MECTA’s devices caused brain damage to the patients. She testified that she was not even curious why multiple people had sued her company for causing them brain damage, assuming the lawsuits to be “frivolous.”
In sworn deposition testimony in 2004, in an unrelated suit, Robin Nicol, was asked if she or anyone from her company had “made any effort to solicit information from persons who have received ECT to see whether or not they have been harmed.” She responded “no … that is not in the purview of our company’s responsibilities.”
DEVICE MAKERS FAILED TO REPORT ADVERSE EVENTS TO THE FDA
We also know from Riera v Somatics, LLC, 2018, that the shock device maker, Somatics LLC, failed to report adverse events to the FDA for over 40 years, contributing to this lack of data.
The duty to “investigate and report” allegations of “serious injury or death” by the manufacturer has resulted in ZERO reported adverse events for over four decades, demonstrating a conscious disregard to comply with reporting obligations.
RIERA V. SOMATICS, LLC, 2018, ECT Shock Treatment Litigation – Case Update June 2019
Page 3 of the 2017 Jose Riera vs Mecta Corporate Somatics LLC complaint:
Despite statutory duties under the Food, Drug and Cosmetic Act (“FDCA”) and directives by the FDA, pursuant to the Medical Device Amendments of 1976 (“MDA”) that ECT device manufacturers report information concerning safety and effectiveness testing for their devices to the FDA, no ECT device manufacturer, including MECTA CORPORATION or SOMATICS, LLC, complied with these statutory obligations.
No ECT manufacturer, including either Defendant, responded to the FDA’s first two orders requiring them to submit safety and effectiveness data by May 28, 1982 and August 14, 1997, respectively. Defendants only responded to a third FDA order, mandated by the Safe Medical Devices Act of 1990 (“SMDA”) requiring Defendants to submit “any information known or otherwise available” about the safety and effectiveness of the device, including adverse safety or effectiveness information.
Defendants’ responses failed to include any information relating to the majority of physiological, psychological, and emotional injuries frequently suffered by those who receive ECT shock treatment. Defendants also grossly understated the incidence of death resulting from ECT. Such a response by Defendants failed to comply with their statutory reporting requirements under the MDA and SMDA.
psychrights.org/states/California/ElectroshockClassAction/1-17.3+–*/0911Complaint.pdf
PATIENTS AND ADVERSE REPORTING
As for patients, few know how to report their injuries to the FDA, or cognitive impairments hinder their efforts.
TOTAL REPORTS

These numbers don’t add up.
I don’t know about you, but the lack of reports stunned me. These are alarmingly low numbers in contrast to Sackiem’s 2 million a year estimate, the numerous reports in the media and research, and reporting mandates for care providers.
IF YOU WERE INJURED BY ECT, YOU CAN CHANGE THIS
Future reporting goals.
We are 8 reports away from exceeding the highest submission year (29 reports in 2020).
You can help us surpass this number by filing your adverse report with the FDA.
Help us reach 50 reports by January 2022.
Reporting Basics
- Reporting ECT injury is important because it makes device regulators aware of how often “serious adverse events” occur. It also helps safety regulators identify previously unknown problems or risks.
- When the FDA receives enough reports, it leads to safety investigations and actions to protect patients from unknowingly risking injury.
- While the form may seem overwhelming, only four questions need to be answered and can be done without your medical file.
- It’s better to report what information you have than none at all.
- MedWatch is the FDA’s system for confidentially reporting adverse medical events from various medical products. Problems with medical devices reported to MedWatch are stored anonymously in a publicly searchable database called Manufacturer and User Facility Device Experience (MAUDE).
- Reporting adverse medical events is voluntary for patients. Many ECT patients and their families are unaware of how to report ECT injuries. Some patients are too injured to participate in reporting injuries without assistance from family, caregivers, mental health professionals, or patient safety advocates.
- Doctors and nurses treating patients with a medical device are obligated by their code of ethics to report serious adverse effects caused by a medical device. Medical professionals treating patients with a history of ECT are also ethically responsible for reporting suspected ECT injury.
- Device manufacturers are legally required to follow up on MAUDE reports to better understand what happened so that risk of future injury can be reduced or eliminated.
- During court depositions taken for the Riera v Somatics, LLC case, The CEO of the Thymatron medical device testified that Somatics, in more than 50 years of making ECT devices, the company had never followed up on any adverse event reports filed with the FDA, willfully negligent of their responsibility to reduce risks and ensure patient safety.
- The successful settlement of Riera v Somatics, LLC proves how not following up on ECT patient reports can be used to demonstrate willful negligence.
- Doctors, ECT providers, mental health counselors, and social workers are bound by their code of ethics to report the suspected harm of individuals with disabilities—even if it was accidental. Failure to report the injury of an individual from a vulnerable population (living with disability) is professional negligence.
NUMBERS Matter
It takes lots of reports to trigger action–- we need a group effort to make a meaningful impact.
We’re already making history.
And we’re on the right track–we have almost surpassed the highest reporting year in just a few months!
WHO CAN REPORT TO FDA
People from all countries can report adverse events to the FDA.
Why non-Americans should report to the FDA.
The FDA influences medicine globally. What the FDA decides impacts the world and, in turn, medical practices in your country. Regardless of your location, the FDA affects your medical care.
- See FDA Goes Global
WHAT KEEPS INJURED ECT PATIENTS FROM REPORTING
Common reasons ECT patients don’t report are privacy concerns, cognitive disability, missing or forgotten information, and trauma.
Let’s break down these barriers:
Fear of retaliation from the device manufacturer or doctors.
MedWatch is anonymous. You can keep your identity private from the device manufacturer (form section: reporter> name & privacy).
The form is confusing.
Yes, the form can be incredibly confusing for former ECT patients. Our guide will walk you through the entire process.
I don’t have all the information they ask for.
The good news is you only need to answer four questions, and the information doesn’t have to be super specific. You don’t have to prove your injuries– only report them.
Remember, it’s better to report a small amount of information than nothing.
What’s the point? The FDA doesn’t care about us.
There’s no doubt the FDA has betrayed the public with how its managed the ECT device:
- Appealing the FDA’s Denial of ECT’s Harms
- Comments by Shock Survivors and Their Loved Ones – Mad In America
- Open Letter of Appeal to FDA about Electroconvulsive Therapy (ECT)
- Open Letter to the FDA’s Denial of ECT’s Harms – 2021
- The FDA’s Regulation of ECT (Shock Treatment): A Beginner (or Refresher) Course
That said, the agency’s behavior shouldn’t be a reason to give up your right to be heard and participate in the regulatory process.
I’ve tried reporting my injuries before– I’m sick of no one listening.
I understand how traumatic trying to get help after ECT is. Knowing the limits of reporting beforehand can make this easier.
It won’t get me the help I need now.
It’s true. Submitting a report doesn’t guarantee immediate or future help for us, but its a crucial part of the process:
- It serves as a stepping stone to investigations and safety actions.
- It’s a way to be seen and heard in a formally recognized way.
- It can lead to beneficial changes for past, present, and future patients.
- Even if the FDA doesn’t follow through, reports stored in the MAUDE database can be used in research and as court evidence.
Speaking truth to power.
As we have seen, ECT device manufacturers have neglected their legal and ethical duties to report adverse events to the FDA. This negligence has contributed to scores of ruined lives. Reporting is just one way to make our injuries known and protect future patients. Even a little effort can go a long way.
HOPE FROM THE PAST; TEN FORMER ECT PATIENTS CHANGE THE WORLD
In 1982, the electroshock device almost got a free pass to class II risk classification.
An ECT survivor named Marilyn Rice rallied ten peers to speak at the FDA reclassification hearings. By speaking out, this small group protected countless patients by ensuring the ECT device stayed in the highest risk category for decades.
“It has been said that it only takes a few people to change the world. Rice had understood this from the beginning. She had worked hard to get survivors to testify at the FDA because she knew that if they did not, the FDA would grant the APAs petition. She was right. After the public hearing ended, when the panel met for closed discussion and to vote, it was clear that some of the panel members were deeply concerned about the testimony from former patients, and could not scientifically or in good conscience reconcile what they had heard with a vote for Class II.”
Doctors of Deception: What They Don’t Want You to Know about Shock Treatment, Linda Andre
This victory shows us what a handful of ECT survivors can pull off when they work through the system.
It’s our turn to stand together and be heard.
HOW TO REPORT
Below are links to our guides for reporting online.
- FDA Reporting Guide – Slideshow, Step by step instructions, links
- FDA Reporting Guide – PDF, Instructions, and side effect list (see updated side effects list here)
- Complete Guide In-depth blog post and all tutorials on adverse reporting
Other ways to report.
- Phone – Call FDA at 1-800-FDA-1088 to report by telephone
- Mail – Download & print paper report
Need more help?
If you still need help reporting we suggest you take our guide to your local librarian; many are trained to help patrons with various tasks. They should be able to help you use a government website.
SUMMARY of Findings
- An estimated 2 million people receive ECT each year, with a 34% increase in American hospitals providing ECT after the 2018 device reclassification.
- The number of annual adverse reports submitted to the FDA is low compared to large numbers of ECT recipients and reported adverse events discussed in case studies, research, and informal reports in the media and patient forums.
- Common serious adverse events like asystole should lead to more reports but have not.
- Deaths and disabling injuries go unattributed to ECT in other ways. Abuses by poorly trained providers happen in absence of treatment standards and enforcement.
- Patients report side effects to their doctors but often aren’t taken seriously, nor do these doctors fulfill their mandated duty to inform the FDA.
- The APA and other psychiatrists advocated against formal neuropsychological evaluations for patients rather than trying to meet the standard, assuring the FDA that nonspecified tests are sufficient.
- Depositions from Robin Nicole, CEO of Mecta, show indifference to those alleging brain damage from their product.
- In Reira v Somatics LLC, it was revealed the device maker Somatics failed to file known adverse reports with the FDA, contributing to a lack of adverse report data.
- Reporting adverse events to the FDA is a way to have your experience heard by a regulatory agency in a way that may lead to safety actions taken to protect future recipients and lead to recognition for those harmed.
- Reporting adverse events through MedWatch benefits people in all countries due to FDA’s global influence.



